Indice Anterior Siguiente
Revista de Ciencias Médicas La Habana 2008; 14 (2)
POLICLÍNICO COMUNITARIO DOCENTE “FLORES BETANCOURT”. ARTEMISA
SINDROME DE TOURAINE-SOLENTE-GOULE. (PAQUIDERMOPERIOSTOSIS PRIMARIA). REPORTE DE UN CASO
Dr. Eduardo Rivero Reyes
Especialista I grado en Dermatología. Profesor Instructor.
RESUMEN
Se presenta un caso de 37 años de edad con diagnóstico de Paquidermoperiostosis primaria o Síndrome de Touraine-Solente-Goulé, afección infrecuente, caracterizada por Paquidermia, Periostosis y Paquidactilia, que puede ser idiopática, con inicio en la pubertad, de origen genético, transmisión autosómica dominante y en el presente caso con una variación fenocópica. En la búsqueda de información realizada no se encontraron reportes previos de esta patología en la literatura cubana, por lo que pudiera tratarse del primer caso reportado en el país.
Descriptores DeCS: OSTEOARTROPATÍA HIPERTRÓFICA PRIMARIA /genética; OSTEOARTROPATÍA HIPERTRÓFICA PRIMARIA /etiología
INTRODUCCIÓN
La Paquidermoperiostosis primaria o idiopática es una genodermatosis poco frecuente, que se define como una osteoartropatía hipertrófica de origen genético, con un carácter autosómico dominante y penetración variable, con predominio en hombres, aunque no se ha encontrado aún el cromosoma responsable.1 Se encuentra historia familiar en el 25- 38% de los casos, la forma primaria de la enfermedad presenta un predominio en hombres con una relación de 9:1, sin ningún tipo de preferencia racial.2
La enfermedad se describió por primera vez en el año 1881 (siglo XIX), caracterizándose posteriormente en el siglo XX, en el año 1935. El inicio de la enfermedad en su forma idiopática ocurre en la pubertad, extendiéndose por un lapso de 5 a 10 años, sin presentarse con posterioridad, más cambios por el resto sus vidas. Se estima que estos cambios puberales sean debidos a coincidir con una tasa de mayor crecimiento en esta etapa 2,3 existiendo informes sobre la alta frecuencia de queloides, en su forma primaria, con presencia de oligofrenia en algunos pacientes.2
La paquidermoperiostosis se divide en dos formas clínicas; una primaria o idiopática que ya se ha mencionado y la forma secundaria cuya etiología es debida a patologías pulmonares, como adenocarcinomas, carcinoma epidermoide bronquial, mesotelioma pleural, abscesos pulmonares; y otras patologías como los carcinomas esofágicos, gástricos, incluso asociada con hipertrofia gástrica, con desarrollo de ulcera gástrica (Enf. de Menetrier), así como también a carcinomas tímicos, osteosarcomas, leucemia mieloide y penfigo paraneoplasico.4-6 La diferencia principal entre la forma primaria y secundaria radica en la edad de presentación, donde la primaria es en la pubertad- juventud, mientras que la secundaria es más frecuente entre los 30 a 50 años de edad.7
La presentación clínica de esta enfermedad, como ya se explicó, se inicia durante la pubertad, entre los 12 a 15 años, con cambios faciales, engrosamiento de la dermis, llamada dermopaquia y un patrón característico en cuero cabelludo, descrito como ¨Cutis Verticis Gyrata¨, que puede también estar presente, aunque no tan ostensible en otras entidades. La piel de manos y pies también está comprometida, constituyendo la llamada Paquiacria. Existe una hiperactividad en las glándulas sebáceas (sebocitosis) y en las sudoríparas (hiperhidrosis) a nivel de toda la piel, siendo más notable en cara, cuero cabelludo, pies y manos. Asimismo, ocurre una periostitis proliferativa en la diáfisis ósea con engrosamiento de los huesos de la tibia, radio y falanges de pies y manos; así como en la bóveda craneal; clínicamente se traduce como un engrosamiento cilíndrico de dedos de pies y manos, llamado paquidactilia, con un aspecto característico de los extremos de estos, denominado dedos hipocráticos, con uñas en vidrio de reloj.
Los tratamientos, actualmente, van dirigidos hacia la resolución de los problemas estéticos de los pacientes.8-10. Dado lo infrecuente de esta entidad, nos propusimos describir el caso y realizar una revisión bibliográfica de esta patología.
PRESENTACIÓN DEL CASO
Paciente masculino de 37 años de edad, casado, de la raza negra, ex deportista, licenciado en Cultura Física, con antecedentes patológicos familiares de alergia (asma y rinitis); ambos padres y antecedentes patológicos personales de bronquitis asmoide, sacrolumbagia y sífilis reciente.
Refiere, que alrededor de los 15 años de edad, comenzó a presentar un aumento marcado de los pliegues en las arrugas de la frente; con aparición de pliegues en la nuca, acompañados de trastornos en la sudoración y la secreción sebácea, así como dolores articulares y aumento ostensible de pies y manos, los cuales notaba eran mayores al resto de sus compañeros, a lo anterior se sumaban uñas algo agrandadas y protuyentes (en vidrio de reloj). Asociándose a lo anterior un aumento de su obesidad, pues recuerda que siempre fue muy obeso.
Desarrollo psicosocial dentro de límites normales, pues manifiesta, que los cambios en su somatotipo, nunca le preocuparon, pues los atribuía a su actividad deportiva (luchador), por lo que nunca procuro orientación médica autorizada al respecto, además de no causarle esto, inconvenientes personales, sociales, ni profesionales. Si notó que todo este proceso cesó hace aproximadamente 10 a 12 años atrás, coincidiendo con su retiro del deporte activo, señalando como único aspecto notable a destacar por ese entonces, la coloración más oscura de las arrugas de la frente, que atribuyó al roce del colchón de lucha.
Al examen físico se observa, la llamada Facie de Angustia de la paquidermoperiostosis, con engrosamiento notable de la piel – demopaquia – con surcos y arrugas profundos a nivel de cuero cabelludo, frente y nuca, donde en esta última, se obsevan surcos gruesos de piel en número de 3 a 4; con un espesor entre 2.5 a 3 cms, constituyendo la llamada ¨cutis verticis gyrata¨,(foto 1) en la frente donde las arrugas adquieren una presentación cerebriforme, con forma el llamado ¨cutis paquidermis frontalis¨este último con una notable hipercromia negro pizarrosa observándose una discreta macroquilia, con paquimenia; moderada hiperhidrosis, más marcada en pies y manos y sebocitosis notable en cara y nuca.
Foto 1
Al examen físico del Sistema Osteromioarticular (SOMA), se observaron como datos positivos, cierta limitación y dolor a la movilización de la articulación de la rodilla y región sacrolumbar, así mismo aumento del tamaño y volumen de pies y manos, paquiacria, con engrosamiento cilíndrico notable de los dedos, paquidactilia. En la piel, a nivel de pies y manos, presenta una hiperqueratosis moderada, la llamada queratodermia palmoplantar del síndrome de Touraine. Solente – Goule. Por último, a nivel de los dedos, se observó un moderado Hipocratismo Digital, con uñas gruesas en vidrio de reloj, paquioniquia con un aumento del ángulo de Lovibond en las mismas.
Con estos datos clínicos se integró el diagnóstico de Paquidermoperiostosis primaria, la cual se caracteriza, por la existencia de tres criterios clínicos mayores o triada clásica: Paquidermia, Periostosis y Paquidactilia.
Para corroborar todo lo anterior se efectuaron estudios de laboratorio, radiológicos, histológicos y psicológicos, encaminados a confirmar los criterios clínicos precedentes y descartar otras entidades nosológicas. Se realizaron, además, estudios básicos de laboratorio Glicemia, P.T.G., Colesterol, Calcio, Fósforo, Hemograma y Conteo de Eosinófilos, con resultados dentro de parámetros normales excepto el hemograma, que arrojó una discreta anemia y los eosinofilos, francamente elevados.
Los estudios radiológicos consistentes en T.A.C para estudio de la silla turca, no muestra alteraciones. Rx de cráneo, engrosamiento moderado del periostio a nivel cefálico, bóveda craneal gruesa, con ausencia de prognatismo y deformaciones faciales (foto 2), Rx de tibia. Proliferación del periostio, donde el contorno del hueso esta duplicado y aumentado el grosor cortical de la diáfisis ósea. Rx de la mano, engrosamiento de los huesos de la falange, con proliferación periostal, que produce pérdida de la curvatura normal epifisiaria, adquiriendo aspectos de ¨cilindros¨.
Foto 2
Biopsia de la piel del cuero cabelludo: Presencia de un epitelio sin lesiones, con hipertrofia de fibras colágenas y aumento de las bandas de colágeno, con fibras de diferente calibre a nivel de la dermis y apéndices cutáneos, con hipertrofia de los complejos pilosebaceos, aumento de la mucina dérmica, el número de fibroblastos y la sustancia cementaría 11(foto 3).
Foto 3
Se practicó un test mental, para medir, nivel de inteligencia al paciente, seleccionándose la Prueba de Kent, modificada 12; la cual mediante un cuestionario de diez preguntas nos brinda tres posibles resultados diagnósticos:
- Deficiente – 19 puntos.
- Normal - 20 a 31 puntos.
- Superdotado + 32 puntos.
El paciente alcanzó calificación de 23 puntos, situándose dentro del parámetro de normalidad; descartando por lo tanto, cualquier tipo de Retraso Mental en el mismo.
DISCUSIÓN
El diagnóstico de esta patología siempre es inicialmente clínico, con base a la triada clásica: Paquidermia, Periostosis y Paquidactilia, constituyendo las manifestaciones mencionadas la forma completa de la enfermedad, basada en los criterios mayores, siendo los criterios menores ¨cutis verticis gyrata¨, hiperhidrosis, sebocitosis, queratodermia palmoplantar, artralgias, anemia, ulceras gástricas, baja talla y retraso mental.2,3 Al mismo tiempo, se pueden identificar, otras dos posibles expresiones de la enfermedad: La forma incompleta, caracterizada por la ausencia de cutis vertici gyrata, y la Forma Fruste caracterizada por dos o mas hallazgos cutáneos, pero sin alteraciones óseas.2-4 Es de señalar que nuestro caso reúne los tres criterios mayores y seis de los criterios menores, lo que lo hacen un caso de Paquidermoperiostosis franco, los criterios menores presentes en cuestión son: ¨cutis verticis gyrata¨, hiperhidrosis, sebocitosis, queratodermia palmoplantar, anemia y artralgias.
La causa fisiopatogénica se desconoce aún, pero se han planteado varias teorías que hablan de un flujo sanguíneo periférico alterado, que provoca éxtasis capilar, dando lugar a hipoxia local y proliferación de colágeno y fibroblastos así como depósito de ácidos mucopolisacáridos y material fibrilar en la dermis 2-4,13,14. Se han estudiado también pacientes con paquidermoperiostotis primaria, con concentraciones altas del factor de crecimiento derivado de plaquetas y de un factor de crecimiento que provoca un estimulo en los fibroblastos de las células endoteliales y los osteoblastos.2-4,15
Por último, también se ha estudiado el papel de los receptores esteroideos de hormonas sexuales y receptores de factores de crecimiento; encontrándose varios de ellos elevados en las enfermedades proliferativas y síndromes paraneoplásicos.6,16
El presente caso, basado en los elementos antes aportados, se enfocó como un Síndrome de Touraine-Solente-Goule, ya que al análisis de los elementos clínicos, se le sumarian los datos complementarios que incluirían elementos genéticos, como el árbol genealógico del paciente (gráfico 1), que aportarían datos esenciales para una mayor comprensión además de un enfoque apropiado, al síndrome en general y a este caso en particular.
Gráfico 1: Pedigrí sobre la Herencia Autosómica Dominante. Variedad Fenocópica del Síndrome de Tourane-Solente-Gole.
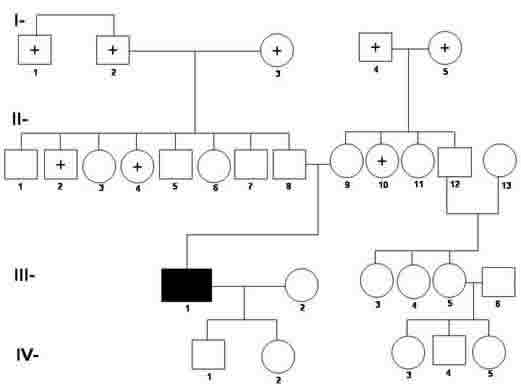
LEYENDA:
Enfermo
Sano
Fallecido
Se conoce que en los casos de herencia autosómica dominante, cada persona afectada tiene por lo menos un progenitor afectado y al casarse con una persona no afectada, tiene como promedio igual número de hijos afectados que sanos, sin importar el sexo, apareciendo el rasgo en cada generación, incluyendo los hijos y nietos de personas sanas, pero con padres afectados.17 En nuestro caso, al padecer de una enfermedad autosómica dominante y no cumplirse este principio de la herencia, como observamos en el Pedigrí; se valoró que se trata de una de las variaciones genéticas de la regla anterior llamada Fenocopias, donde fenotípicamente existe la enfermedad, pero no existe un gen mutante reciente como ocurre en las mutaciones, pues no hay descendencia afectada, y que mas bien parece depender de un genotipo algo diferente al mutante dominante, pues no existen, como en este paciente, antecedentes familiares de enfermedad, pero tampoco transmisión de la misma a su descendencia17. Por lo que inferimos que estamos ante un caso de herencia dominante autosómica, variante fenocópica de la enfermedad Paquidermoperiostosis primaria.
Con respecto a otros posibles diagnósticos, por demás, entidades nosológicas, bastante infrecuentes, tenemos en primer lugar a descartar la Acromegalia, clínicamente la descartamos por la no presencia en el paciente de deformaciones del macizo facial, ni prognatismo; en las extremidades, aunque en ambas entidades existen aumentos de volumen considerables, en la acromegalia, existen uñas pequeñas, surcadas por estrías, todo lo contrario de la Paquidermoperiostosis donde las uñas son grandes en vidrio de reloj, además, la piel a este nivel tiene un engrosamiento notable, dermopaquia, ausente en la acromegalia. Súmese a lo anterior la curva de tolerancia a la glucosa que presenta un comportamiento dentro de los parámetros establecidos y no existiendo una Hiperfosfatemia, elementos estos que nos sirven para eliminar definitivamente este diagnóstico en nuestro caso.
En la Acropaquiatiroidea el crecimiento se da en las manos y pies solamente, paquiacria, asociado a exoftalmos, sin otras alteraciones de esta índole en el resto del organismo, como si ocurre en la Paquidermoperiostotis; las alteraciones cutáneas son de tipo mixedematoso como en el hipertiroidismo, sin engrosamiento cutáneo, por último se puede observar en estos casos una hipocolesteloremia, asociada a otros elementos de hipertiroidismo, que no están presentes en este caso.
Otro diagnóstico posible a descartar en el Síndrome de Rosenthal - Klofpfer que está caracterizado por un aumento del macizo facial, pero sin alteraciones de la silla turca, siendo la primera de estas dos características, la no presente en nuestro paciente; en esta patología a nivel de piel, si se presenta, el ¨cutis verticis gyrata¨, pero no tiene la magnitud de la Paquidermoperiostoasis, además de no presentar, la acropaquia, paquidactilia y periostosis. En el aspecto genético del Síndrome de Rosenthal - Klofpfer se define como una afección genética autosómica dominante, pero a diferencia de la Paquidermoperiostosis donde puede existir retardo mental en el Síndrome de Rosenthal - Klofpfer nunca esto es parte del cuadro clínico por lo que unido a lo ya antes referido sirve para descartar la entidad, en nuestro caso.
Por último, otros diagnósticos también posibles a descartar serían: Síndrome de Soto o Gigantismo Cerebral y el Síndrome de Brugsh o Acropaquidermia.
El primero de los síndromes es una afección genética extremadamente rara que se caracteriza por aumento de talla y peso, con prognatismo y facciones acromegálicas, con engrosamiento de pies y manos, clínicamente al no existir prognatismo, ni fascie acromegálica, unido a un engrosamiento no tan solo limitado a pies y manos sino que abarca toda la piel, uñas y periostio, nos permiten descartarla clínicamente, sumándose a esto la siempre presencia de retardo mental en los pacientes aquejados de esta rara enfermedad lo que no siempre se cumple en la Paquidermoperiostotis como en este caso, nos hace eliminarla definitivamente.
La otra patología, la Acropaquidermia o Síndrome de Brogsh, presenta engrosamientos de pies y manos con dedos en masa, pero sin alteraciones a nivel cefálico, tan solo una hipertrofia a nivel de huesos largos, con un engrosamiento cutáneo que se circunscribe a nivel acral solamente. Metabólicamente, cursa con una disminución del calcio en sangre, por aumento de la acreción cálcica; todos estos elementos antes referidos sirven para descartar la entidad, pues en la Paquidermoperiostotis, la periostosis es más general y no tan solo acral, además de ser el engrosamiento cutáneo generalizado, con niveles de calcio en sangre dentro de limites normales; por todo lo anterior, no es, considerada finalmente, la Acropaquidermia.4,18-23
Tabla : Sobre las principales entidades nosológicas a descartar.
ENFERMEDADES |
EXAMENES COMPLEMENTARIOS. |
||||||||||
Glicemia |
Calcio |
Fósforo |
Colesterol |
Hemograma |
Alteraciones |
Alteraciones |
Alteraciones |
Retraso |
|||
SOMA |
|||||||||||
C |
MF |
E |
|||||||||
Acromegalia |
(+) |
(+) |
(+) |
(+) |
(+) |
(+) |
|||||
Acropaquia Tiroidéa. |
(-) |
(+) |
(+) |
||||||||
Síndrome de Rosenthal – Kloepfer. |
(+) |
(+) |
(+) |
||||||||
Síndrome de Soto Gigantismo Cerebral. |
(+) |
(+) |
(+) |
(+) |
|||||||
Síndrome de Brugsh Acropaquidermia |
(-) |
(+) |
(+) |
||||||||
Síndrome de Touraine-Solente-Goule |
(-) |
(+) |
(+) |
(+) |
(+) |
(+/-) |
|||||
Fuente: Datos de Laboratorio clínico
LEYENDA:
Normal o ausente.
Disminuido.
Elevado o presente.
En cuanto a su pronóstico, el mismo es bueno, ya que la enfermedad se estabiliza después de 10 a 12 años, del inicio de la misma debido quizás a una taza de crecimiento, ya no tal alta como en la adolescencia, recomendándose en este momento de quiescencia realizar los tratamiento quirúrgicos estéticos para mejorar la apariencia del paciente.8,9,18-23
A manera de conclusión se puede señalar que el caso descrito se enmarca, tanto desde el punto de vista clínico como en el de los exámenes complementarios, dentro de los parámetros establecidos internacionalmente para esta enfermedad y que se comprueba en este caso. Lo realmente interesante en este caso es el hecho de tratarse de una forma completa de Paquidermoperiostosis primaria con una variación fenocópica de su herencia autosómica dominante y ser el primer caso que se reporta en nuestro país, según la literatura consultada.
REFERENCIAS BIBLIOGRÁFICAS
- Rimoin DL. Pachydermoperiostosis primary oridiopatmic hipertropmic osteoarthropaty. Am J Med 1962; 33: 166 – 87.
- Correa Carrillo M. Osteoartropatía hipertrofica primaria, paquidermoperiostosis. Rev Mexicana 2003; 47(4): 106 - 109
- Touraine A, Solente G, Goulé L. Un síndrome osteodermopathique: la pachydermie plicaturee avec pachy periostose des extremites. Presse Med 1935 ; 43 : 1820-4.
- Champion Rook MW. Englies: textbook of dermatology (monografía en CD-ROOM). Versión 1.2.0 Software Gentian. London: Blanckwell Sciencie; 1998.
- Shimizo C. Ararecase of acromegal y associated witm pachydermoperiostosis. J Endodrinol Invest 1999; 14: 181-2.
- Lam S, Stone MS. Paraneoplastie pempehiaus, cicatricial conjunctivitis andacantosis niaricans with pachydermotoglypmy in patient witm squamous ell carcinoma. Oftmalmology. 1992; 99:108-13.
- Benjamín Hidalgo M, Norman Hines J. Síndrome de Touraine- Solente- Goulé : breve revisión y reporte del primer caso en Costa Rica. Rev Costarricense de Ciencias Méd 2001; 22 (3-4): 119 – 26
- González Saldivar G. Síndrome de Tooraide-Solente-Goule: presentación de un caso clínico. Dermatología. 1997; 7: 9-11.
- Omtsokam Takayanagis E. Reconstruction in pachydermoperiostosis. Plast Reconstr 1988; 81: 88 – 90.
- Friedmofer M, Salles AG. Correction of eyelid anormalies in pachydermperiostosis. Opmtmal Plast Reconstr Sur 1999; 15: 137-8.
- Elder D. Lever s histopathology ortheskin lippincot. 8ed. Boston: Raven; 1997.
- Universidad La Habana, Departamento de Psiquiatría y Psicología Médica. Psiquiatría. Ciudad de La Habana: Pueblo y Educación; 1974. T 2.p.52-3.
- Heddayati M, Barmada R, Skosey TL. Acrolysis in pachydermoperiostosis: primary idiomathic hypertropmie osteoartrhropathy. Arch Intern Med 1980; 140: 1087-8.
- Olkarinen A, Palatsi R. Pachydermoperiostosis analysis of the connective tissue absnormality in one family. J Am Acad Dermatology 1994; 31(6):947-53.
- Vogl A, Goldfischer S. Pachydermoperiostosis primaryor idiopathic hypertrophic osteoartropathy. Am J Med 1962; 33: 166-87.
- Martínez -Lavin M. Primary hypertrophieosteoarthropathy; another disorder associated with patent doctus arteriosus. Pediatr Cardiol 1993; 14: 181-2.
- Berkow R. El manual Merck de diagnóstico y terapéutica. 7 ed. México, DF: Nueva Editorial Interamericana; 1986.
- Domonkos Andrews A. Tratado de dermatología. La Habana: Científico Técnica; 1984.
- Diccionario Terminológico de Ciencias Médicas. 10 ed. Barcelona: Salvat; 1972; Paquidermoperiostotis.
- Castori M, Sinibaldi L, Mingarelli R, Lachman RS, Rimoin DL, Dallapiccola B, et al. Pachydermoperiostosis: an update. Clin Genet 2005; 68: 477–86.
- Amello V, Francisca Barbiri H, Marco Valenzuela H. Paquidermoperiostosis u Osteoartropatia Hipertrofica Idiopatica (caso radiológico). Rev Chil Radiolog 2001; 7(2): 40- 74.
- Karkucak M, Erturk E, Akyazih. Primary hipertrofic osteoarthropat. (Pachidermoperiostosis) a case report. Rheumatol Int 2007; 27(4): 403-5.
- Kabi F, Mkinsi O, JananiS Raissouni N. Pachydermoperiostosis: a case report. Rev Med Interne 2006; 27(9): 710-2.
SUMMARY
A 37-year-old case is presented with diagnosis of Primary pachydermoperiostosis or Touraine-Solente-Golé syndrome, an infrequent affection, characterized by Pachydermia, Periostosis and Pachydactylia, that can be idiopathic, starting in puberty, and of genetic origin, dominant autosomic transmission and in the present case with a phenocopic variation. In the search for information that was carried out no prior reports of this pathology were found in the Cuban literature, so it is possible this could be the first case reported in the country.
Subject Headings: OSTEOARTHROPATHY, PRIMARY HYPERTROPHIC /genetics; OSTEOARTHROPATHY, PRIMARY HYPERTROPHIC /etiology
Dr. Eduardo Rivero Reyes
Calle Manuel Valdés no. 4810, entre República y Martí.
Artemisa, CP-33800, La Habana, CUBA.