Anterior Indice Siguiente
Revista de Ciencias Médicas La Habana 2015; 21(2)
PRESENTACIÓN DE CASO
Elayne Esther Santana HernándezI, Víctor Jesús Tamayo ChangII.
IEspecialista de I y II Grado en Medicina General Integral y Genética Médica. Máster en Atención Integral al Niño. Investigador Agregado. Profesor Asistente.
Centro Provincial de Genética Médica. Holguín, Cuba. Correo electrónico: esantana@hpuh.hlg.sld.cu
IIEspecialista de I y II Grado en Genética Médica. Máster en Atención Integral al Niño. Investigador Agregado. Profesor Auxiliar. Centro Provincial de Genética Médica. Holguín, Cuba. Correo electrónico: vtamayoa@hpuh.hlg.sld.cu
RESUMEN
El Síndrome Treacher-Collins es un trastorno craneofacial congénito que se produce por la mutación del gen TCOF1, localizado en el cromosoma 5q31-33. Se caracteriza por malformaciones mandibulofaciales, donde se destacan la micrognatia severa, macrostomia y microtia de grado variable. La forma de herencia predominante es la autosómico dominante, aunque se han reportado casos autosómicos recesivos y mutaciones de novo. Se presenta este caso teniendo en cuenta lo poco frecuente de este síndrome malformativo, con el objetivo de destacar la importancia del método clínico como forma de diagnóstico eficaz, al presentarse un recién nacido con malformaciones severas es necesario diagnosticar oportunamente la afección que presenta para ejecutar intervención multidisciplinaria que requiera y brindar un adecuado asesoramiento genético a la familia.
Palabras claves: disostosis mandibulofacial; anomalías craneofacial; cigoma.
ABSTRACT
Treacher-Collins Syndrome is a craneal facial congenital disorder that is produced by the mutation of the TCOF1 gen, localized in the 5q31-33 chromosome. It is characterized by mandibular facial malformations, where severe micrognaty, macrostomy and microty of variable grade are relevant. The predominant inherited way is the dominant autonomics, although autonomics recessive cases and novo mutations have been reported. This case is presented taking into consideration that this malformation syndrome is not frequent, with the objective to emphasize the importance of the clinical method as a good diagnostic way, when a newborn was presented with severe malformations it is necessary to diagnose the affection that he/she presents early to execute the required multidisciplinary intervention and give an adequate genetic recommendation to the family.
Keywords: mandibulofacial dysostosis; craniofacial abnormalities; zygoma.
INTRODUCCION
El síndrome Treacher Collins (STC) fue descrito inicialmente en 1846 por Thompson y posteriormente, por Berry en 1889.1,2 Fue el oftalmólogo inglés E. Treacher Collins quien describió sus características principales en 1900.3-5 Después Franceschetti y Klein fueron los primeros en usar la expresión disostosis mandibulofacialy describieron el perfil de los afectados como similar a la cara de los peces o los pájaros.6,7 Su incidencia se estima 1 de cada 25.000 a 50.000 nacidos vivos 8. Se plantea que este síndrome tiene una expresividad muy variable, siendo localizado el gen TCOF1 en el cromosoma 5q31-33, donde se codifica la proteína treacle responsable de las malformaciones, por alteraciones de los dos primeros arcos braquiales.8,9
Las alteraciones faciales básicas que se describen son; hendiduras palpebrales inclinación hacia abajo muy acentuadas se reporta en (89 %), hipoplasia malar en (81 %), micro-retrognatia también extrema en (78 %), característica hendidura en el parpado inferior en (63 %), en cuyo nivel faltan las pestañas en (69 %), y microtia bilateral de grado variable, como las demás anomalías.6-9
Se presenta este caso teniendo en cuenta lo poco frecuente de este síndrome malformativo y considerando lo importante que resulta realizar un diagnostico clínico en etapas tempranas para decidir un seguimiento y tratamiento por un equipo multidisciplinario que evalué por etapas cada afectación. Diagnostico clínico que en ocasiones resulta muy difícil por la expresividad variable de las malformaciones craneofaciales y del cigomático, así como de los pabellones auriculares, donde en muchos casos se han presentado malformaciones graves que ponen el peligro las vías áreas y con ellos la vida de estos afectados.
En Cuba no se dispone del estudio molecular para su confirmación dada la baja incidencia de este síndrome y lo costoso de dicha técnica. Considerando importante el método clínico, que permite una correcta delineación del fenotipo; lográndose el diagnóstico clínico del síndrome malformativo y con esto se puede ayudar a las familias, brindándole un adecuado asesoramiento genético. Considerándose necesario que los afectados sean evaluados oportunamente por las especialidades que lo requieran para su mejor seguimiento y tratamiento.
PRESENTACION DE CASO
Paciente masculino APG de 8 meses de edad.
Antecedentes patológicos familiares: no se recogen antecedentes de matrimonios consanguíneos ni de ningún ancestro en común. No refieren deformidades ni malformaciones mandibulofaciales por vía materna ni paterna. Madre de 23 años de edad, primigesta, HO E2P1A1 (regulación menstrual).
Antecedentes prenatales: las medidas de proporciones faciales eran normales no se visualizaron deformidades en las orejas, los ultrasonidos genéticos y sus mediciones estuvieron en parámetros normales para la edad gestacional.
Antecedentes perinatales: parto eutócico a las 39,5 semanas, peso 2900 gramos; talla 50 cm; apgar 8-9.
Examen físico: deformidad facial al nacimiento dado por pobre desarrollo malar y cigomático, hendiduras palpebrales hacia abajo.
En la neonatología de la maternidad provincial se recibe un recién nacido masculino con malformación congénita craneofacial, se valora en conjunto por neonatólogos y especialista en genética clínica y por el examen físico se realiza el diagnostico clínico de que el afectado presenta el síndrome Treacher-Collins, se sugiere la valoración de oftalmología, otorrinolaringología, maxilofacial.
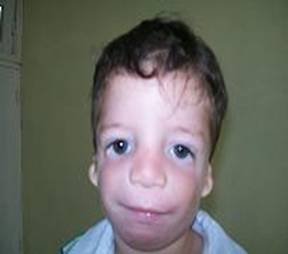
Al examen físico se aprecia; los párpados inferiores presentan una pequeña muesca al nivel de la unión de los dos tercios medianos con el externo, así como escasa presencia de pestañas. El eje de la fisura palpebral está oblicuamente inclinado hacia abajo y afuera, produciendo la ilusión óptica de que existe una brida que tira de ellos hacia abajo a partir de la muesca. Escaso desarrollo malar y cigomático, con microretrognatia severa, filtrum largo con labios muy finos, evidente macrostomía como se observa en la figura 1.
Figura 1. Características faciales del afectado
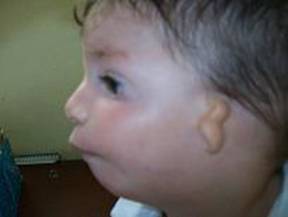
En la figura 2 se aprecia pobre desarrollo de la mandíbula inferior como esta retraída hacia atrás y el rudimentario pabellón auricular, diagnosticándose la microtia grado IV; que se presenta de forma bilateral, con solo esbozo de los pabellones auriculares sin presencia de los conductos auditivos externos.
Figura 2. Vista de perfil del lactante que permite evaluar retromicrognatia y el grado de microtia que presenta.
DISCUSION
El STC es poco frecuente a pesar de que se describe en la mayoría de los casos publicados un patrón de herencia autosómico dominante con expresividad muy variable y penetrancia completa, en el caso que se presenta después de examinar a los familiares de primer y segundo grado de parentesco, se reconoce este nuevo afectado, como una mutación nueva, es decir de novo.1-3
Por lo general, las características faciales del STC son bilaterales y relativamente simétricas. La hipoplasia cigomática y de los rebordes orbitarios hace que haya una aparente protrusión centrofacial, aunque las alteraciones en la región nasal no sean tan graves.4 Las alteraciones del pabellón auricular, la retrognatia, los pliegues palpebrales antimongoloides y micrognatia también son típicas.5 En el cráneo, se describe una disminución de la distancia intratemporal que se plantea que puede ser diagnosticado desde la etapa prenatal.5,6 Hay una convexidad mandibular con macrostomía y secundaria al retrognatismo, que también se puede hacer diagnostico en casos más severos evidenciarse en los ultrasonidos prenatales.6
Con mayor frecuencia, el pabellón auricular presenta microtia de distintos grados que se correlaciona con la gravedad de la pérdida auditiva. También se ha descrito la presencia de apéndices auriculares y alteraciones en el oído medio. Las alteraciones en el oído interno, aunque exóticas, están descritas. Las alteraciones cognoscitivas no son tan frecuentes y, por lo general, se asocian a la hipoacusia.7,8
El diagnóstico del STC es clínico y el manejo requiere una aproximación interdisciplinaria. Se han propuesto varios protocolos de manejo para esta patología, no existiendo un protocolo de seguimiento establecido sin una propuesta concreta, por ser un trastorno infrecuente.9,10
En la provincia con tres casos similares a este se estableció que un equipo multidisciplinario formado por oftalmólogos, otorrinos, maxilofaciales, cirujanos estéticos y genetistas clínicos evaluaran a los afectados y sus familiares cada tres meses en la etapa de lactantes los menos afectados y los mas severos mensual por las posibles complicaciones. A los transicionales cada 6 meses hasta los tres años, cuando se practica la cirugía reconstructiva, lo que realizo a uno con mayor afectación facial e igual malformación auricular.
Hasta el momento se han obtenido buenos resultados clínicos, estéticos y funcionales; brindando un adecuado asesoramiento genético en estas tres familias sin recurrencia del síndrome en ningún otro miembro de estas familias, igualmente se describe en otros casos la experiencia de cirugía reconstructiva que ha mejorado estéticamente elevando su autoestima y mejorando su incorporación social.
REFERENCIAS BIBLIOGRAFICAS
Recibido: 15 de mayo del 2015
Aprobado: 9 de junio del 2015
Dra. Elayne Esther Santana Hernández. Especialista de I y II Grado en Medicina General Integral y Genética Médica. Máster en Atención Integral al Niño. Investigador Agregado. Profesor Asistente. Centro Provincial de Genética Médica. Holguín, Cuba. Correo electrónico: esantana@hpuh.hlg.sld.cu